All Canopy commands are run through ‘canopy’ script that you should already have installed (see the installation instructions).
Contents
Getting help
Setting options
Other options
- Codon-based alignment
- Translated alignment
- Multilocus alignment and species tree inference
- Output formats
- Other software supported by Canopy
Example analysis with Canopy
Inspection of results with Wasabi
Getting help
To see all the options and their explanations use
canopy --help
Setting options
There are three ways to run the tool. The first way is to define all options in a config file:
canopy example/example.cfg
Directory “example” in the downloaded source code provides config files and test data to experiment with the tool.
The second way is to specify options in the command line:
canopy --seq=example/example.fas
Finally, one can combine the two and reset some options while reading other options from a config file:
canopy example/example.cfg --max_iter=2
In the following, the usage of the tool will be demonstrated using the combination of a config file and command line options.
Other options
Codon-based alignment
Canopy can align protein-coding DNA sequences in codon space. If the input is DNA sequences and align_datatype is set to ‘CODON’, canopy performs codon alignment.
canopy example/example.cfg --align_datatype=CODON
Translated alignment
Canopy can translate protein-coding DNA sequences into protein and then align them in protein space. This is done by setting align_datatype to ‘PROTEIN’. The final alignment will be translated back to DNA.
canopy example/example.cfg --align_datatype=PROTEIN
Multilocus alignment and species tree inference
If seq option points to a directory with multiple sequence files (assumed to be different loci for an overlapping set of species), canopy will infer the alignment for each file separately and then estimate a single tree from a concatenated alignment.
canopy example/example.cfg --seq=example/multilocus/
Output formats
By default, the input data, the results of the initial step and the final result pair are saved. More information (including all intermediate files) can be outputted with --save_option=<str> (simple, rich, all). By default, the output alignment files are in FASTA format but this can be changed with option --output_format=<str> (phylip, fasta, nexus). The default format for output trees is NEWICK and it can be changed to NEXUS with option --tree_format=nexus.
In addition to the alignment and corresponding RAxML estimated tree, the guide tree for PRANK-generated alignments will be outputted. The following PRANK options are also supported:
- showxml: output the results in PRANK’s own XML format
- showanc: output the guide tree and the estimated ancestral sequences at each internal node
- showevents: output events happened at each branch
- showall: output all above information
If xml output is selected, the final phylogenetic tree (e.g. from RAxML) is also outputted together with the underlying alignment in file result_final.xml. Note that the phylogenetic tree is unrooted and further analysis of that tree-alignment pair within Wasabi (e.g. ancestral reconstruction) may not be meaningful unless the tree is rerooted.
Other software supported by Canopy
In addition to PRANK and PAGAN, Canopy also supports MAFFT and ClustalW for the sequence alignment step and MUSCLE for the alignment merging. For the phylogenetic inference step, Canopy supports RAxML, FastTree and PhyML.
Alternative tools can be defined with command-line options. The defaults values (after each option) and alternatives (in square brackets) are:
--initial_aligner=mafft [mafft, prank, clustalw, pagan]
--aligner=prank [prank, mafft, clustalw, pagan]
--merger=prank [prank, muscle]
--tree_estimator=raxml [raxml, fasttree, phyml]
Example analysis with Canopy
The config file distributed with the tool looks like this:
[main] seq=example/example.fas work_directory=results/ name=first_run max_prob_size=20 max_iter=1 num_cpus=2 showxml=True decomposite_strategy=seed config_export_path=config.bac initial_aligner=mafft aligner=prank tree_estimator=raxml merger=prank [raxml] args=-p 111111 [prank] args=+F #args=+F #-seed=11 -reproducible
Some arguments re-define the default value for the option and only shown for illustration. The line starting with # is ignored.
When executed with command
canopy example/example.cfg
Canopy runs a simple analysis for the data set example/example.fas and places the results in the directory results/first_run/.
The output files are the following:
- config.bac: config file with parameters used for the analysis
- first_run.log: log file
- input.fas: input data
- initial.fas: initial alignment generated with MAFFT
- initial.tree: initial tree estimated with RAxML
- initial_score.txt: ML score of the initial alignment+tree pair
- result.fas: final output alignment generated with PRANK
- result.dnd: guide tree used for the final alignment
- result.xml: final alignment and its guide tree in XML format
- result.tre: final ML tree estimated with RAxML
- result_score.txt: ML score of the final alignment+tree pair
- result_final.xml: final alignment and final ML tree in XML format
The analysis can be repeated with command canopy config.bac. If the PRANK options -seed=XX -reproducible were specified, the output would be identical.
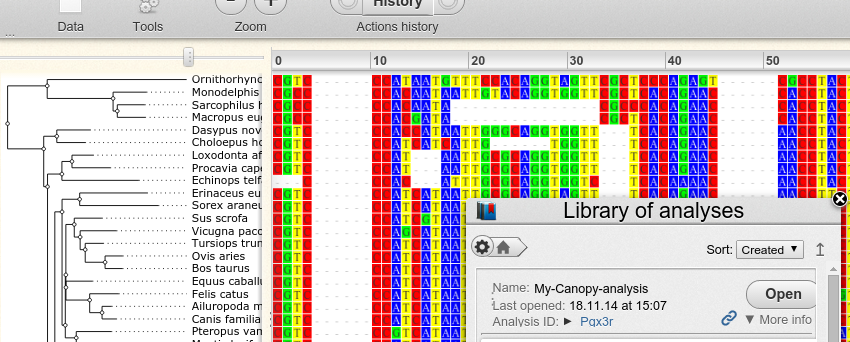
Inspection of results with Wasabi
The xml file written by Canopy can be visualized with Wasabi. To do that, one first has to create an account at Wasabi. The user ID is then the last six characters of the URL:
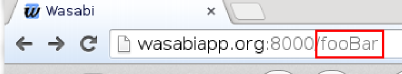
Assuming user ID ‘fooBar’, one can upload a file called ‘result.xml’ to Wasabi with the command:
canopy wasabi fooBar analysis/result.xml My-Canopy-analysis
The three parameters of the command are the user ID, the path to the xml file and a name for the analysis within Wasabi.
With this command the file is exported to Wasabi and a browser window is launched to display it: